
Medwisdom Lifesciences Pvt Ltd provides affordable eCTD publishing services in India with precision, speed, and regulatory compliance at the core. Whether you need end-to-end dossier preparation or targeted eCTD publishing support, our team of experienced regulatory professionals delivers submission-ready packages aligned with FDA, EMA, CDSCO, GCC, and other global authority requirements.
As a growing pharmaceutical company in India, cost-effective regulatory strategy is essential. We combine deep regulatory expertise with competitive pricing — making world-class eCTD compliance accessible to Indian pharma manufacturers of all sizes.
Our end-to-end regulatory dossier solutions are designed to reduce submission timelines, minimize deficiencies, and accelerate product approvals in India and international markets.
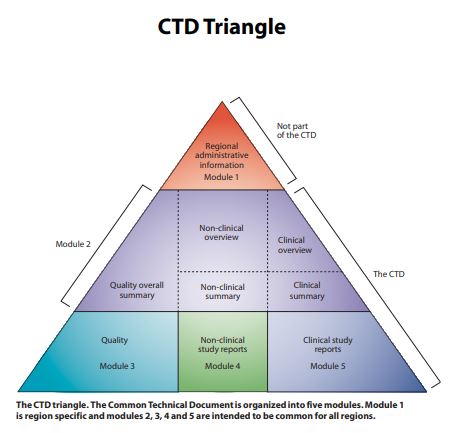
Complete Common Technical Documents (Modules 1–5) tailored for LATAM, Africa, Europe, and Asia-Pacific markets — compliant with ICH M4 guidelines.
Expert eCTD compilation, technical file validation, NeeS formatting, and seamless electronic submission to FDA, EMA, CDSCO, GCC, and beyond.
ASEAN Common Technical Documents for Southeast Asian markets including Malaysia, Thailand, Philippines, Indonesia, and Vietnam regulatory submissions.
Confidential DMFs for APIs, excipients, and packaging materials supporting product registrations with complete manufacturing and stability data.
Variations, supplements, renewals, and post-approval changes managed efficiently to maintain compliance throughout your product's lifecycle.
Comprehensive review of your existing dossier against target market requirements — identifying gaps and creating a clear action plan for remediation.
Our regulatory specialists are proficient across all major dossier formats accepted by global health authorities, ensuring your submissions meet country-specific and ICH standards.
| Format | Full Name | Applicable Regions | Key Authorities | Status |
|---|---|---|---|---|
| eCTD | Electronic Common Technical Document | USA, EU, Japan, Canada, Australia | FDA, EMA, PMDA, Health Canada | Mandatory |
| CTD | Common Technical Document (Paper) | India, Middle East, Africa, LATAM | CDSCO, MOH, NAFDAC, ANVISA | Accepted |
| ACTD | ASEAN Common Technical Document | Malaysia, Thailand, Philippines, Vietnam | NPRA, FDA-TH, FDA-PH, DAV | Required |
| NeeS | Non-eCTD Electronic Submission | European Union (transition) | EMA, National Agencies | Transitional |
| DMF | Drug Master File | Global | FDA, EMA, CDSCO | Supported |
A structured, transparent workflow designed to deliver submission-ready dossiers on time and within budget — every time.
We evaluate your product, target markets, and data availability to define the regulatory pathway and dossier scope.
Our experts identify data gaps, missing studies, and formatting issues in your existing documentation against target authority requirements.
We compile, write, and format all modules — ensuring scientific accuracy, regulatory compliance, and authority-specific requirements.
Final validation, technical QC, eCTD publishing, and electronic or paper submission to the target regulatory authority.
Delivering affordable, compliant, and on-time eCTD publishing services across India and globally since our founding.
Transparent, competitive pricing with no hidden charges — making global-standard eCTD compliance accessible for Indian generics and specialty pharma companies.
Our specialists bring hands-on experience with FDA, EMA, CDSCO, GCC submissions — with proven expertise in eCTD lifecycle management and technical validation.
Streamlined workflows and dedicated project managers ensure your dossier is prepared, validated, and submitted without unnecessary delays.
Multi-level QC review, technical eCTD validation, and compliance verification before every submission — minimizing deficiency notices.
Quick answers to the most common questions about our affordable eCTD publishing services in India.
eCTD (Electronic Common Technical Document) is a standardised, structured format for electronic regulatory submissions to global health authorities. It is mandatory for submissions to the US FDA, EMA (European Medicines Agency), Health Canada, and several other major agencies. eCTD ensures consistent document organisation, version control, and lifecycle management, reducing submission errors and accelerating review timelines.
Our eCTD publishing costs are highly competitive compared to global CROs and regulatory agencies. Pricing depends on the dossier scope, modules required, product complexity, and target regulatory authority. We offer flexible pricing models — per module, full dossier, or retainer — tailored to the budget of Indian pharma companies. Contact us for a personalised, no-obligation quote.
We have experience submitting dossiers to the US FDA, European Medicines Agency (EMA), CDSCO (India), GCC countries (Saudi Arabia, UAE, Kuwait, Qatar, Oman, Bahrain), Health Canada, TGA (Australia), PMDA (Japan), and numerous African and LATAM national authorities. Our global reach ensures your product gets approved in your target market.
We typically need your product's R&D data, analytical study reports, stability data, manufacturing details, clinical/bioequivalence study reports (where applicable), and administrative information. We conduct a gap analysis first to identify missing data and advise on bridging studies needed. The exact document checklist varies by product type and target authority.
Timeline depends on the product complexity and scope. A standard generic product CTD/eCTD typically takes 4–8 weeks for preparation, provided all source data is available. Complex biologics, NCEs, or combination products may take 3–6 months. We provide an estimated project plan after the initial consultation and gap analysis.
Yes. We provide comprehensive post-approval support including Type I/II variations, supplements, label updates, renewals, and eCTD sequence management. Our team ensures your approved dossier remains up to date, compliant, and properly maintained in the eCTD lifecycle throughout your product's commercial life.